氨(NH3)作为重要的化工原料和零碳氢能载体,其规模化应用带来的泄漏风险日益凸显。逃逸的氨气不仅引发呼吸系统疾病、刺激皮肤,还会促进大气颗粒物和二次无机气溶胶的形成,加剧空气污染。选择性催化氧化技术(NH3-SCO)可将氨转化为氮气和水,是理想的末端治理手段。然而现有催化剂存在显著的局限性:贵金属催化剂(如铂基材料)虽在低温下活性优异,但氮气选择性普遍低于50%,易生成有害的氮氧化物副产物;过渡金属氧化物(如铜氧化物)虽选择性较高,却需300°C以上高温才能发挥作用。近年来,铂-铜双金属体系因协同效应而备受关注,但传统制备方法难以实现活性组分的原子级分散与强相互作用,导致金属迁移烧结、稳定性差等问题。虽然核壳结构等空间隔离策略可抑制烧结,却牺牲了双金属界面电子协同这一关键要素。如何在保持原子尺度金属接触的同时实现空间限域与热力学稳定,成为该领域亟待突破的瓶颈。
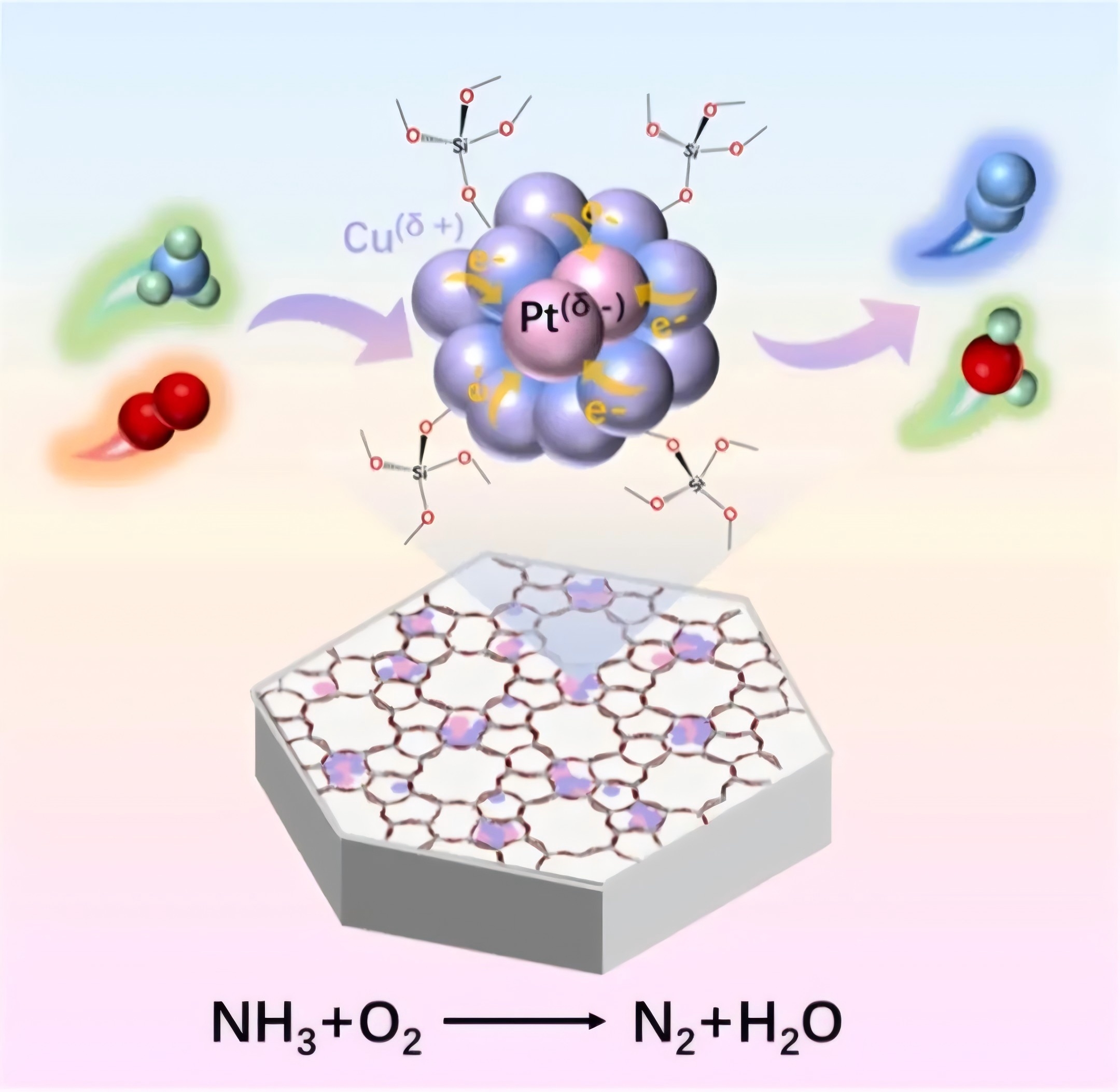
在这项研究中,研究人员通过一锅水热法将亚纳米尺度的PtCu双金属团簇原位封装于纯硅MFI型分子筛(S-1)的孔道结构中,制备出PtCu@S-1催化剂。利用原子分辨率的像差校正高角环形暗场扫描透射电镜(HAADF-STEM)与集成差分相位对比成像(iDPC-STEM)技术,研究团队在[010]方向上直接观测到0.4-0.6nm的双金属团簇选择性占据分子筛的六元环与五元环孔道。同步辐射X射线吸收谱(XAS)与X射线光电子能谱(XPS)分析证实,铜向铂发生定向电子转移,形成极化的Cu(δ+)-Pt(δ-)界面结构。该电子重排使铂的d带中心上调,显著增强氧气解离能力;同时铜的缺电子状态强化了对氨的化学吸附。催化性能评价显示,PtCu@S-1在150°C时的氨氧化速率比传统浸渍法制备的PtCu/S-1催化剂高13倍,活化能降低26 kJ/mol;在250°C时氮气选择性达92%,且在275°C、3%水汽条件下连续运行54小时活性保持稳定。氧同位素交换实验表明,PtCu界面在300°C以上即展现出高效的氧气活化能力,而单金属催化剂需400°C以上才表现类似活性。原位红外光谱进一步揭示了NH3在Cu(δ+)位点吸附,并在相邻Pt(δ-)位点协同作用下逐步脱氢生成NH2和NH中间体的反应路径,最终通过直接偶联或N-O中间体路径生成氮气。
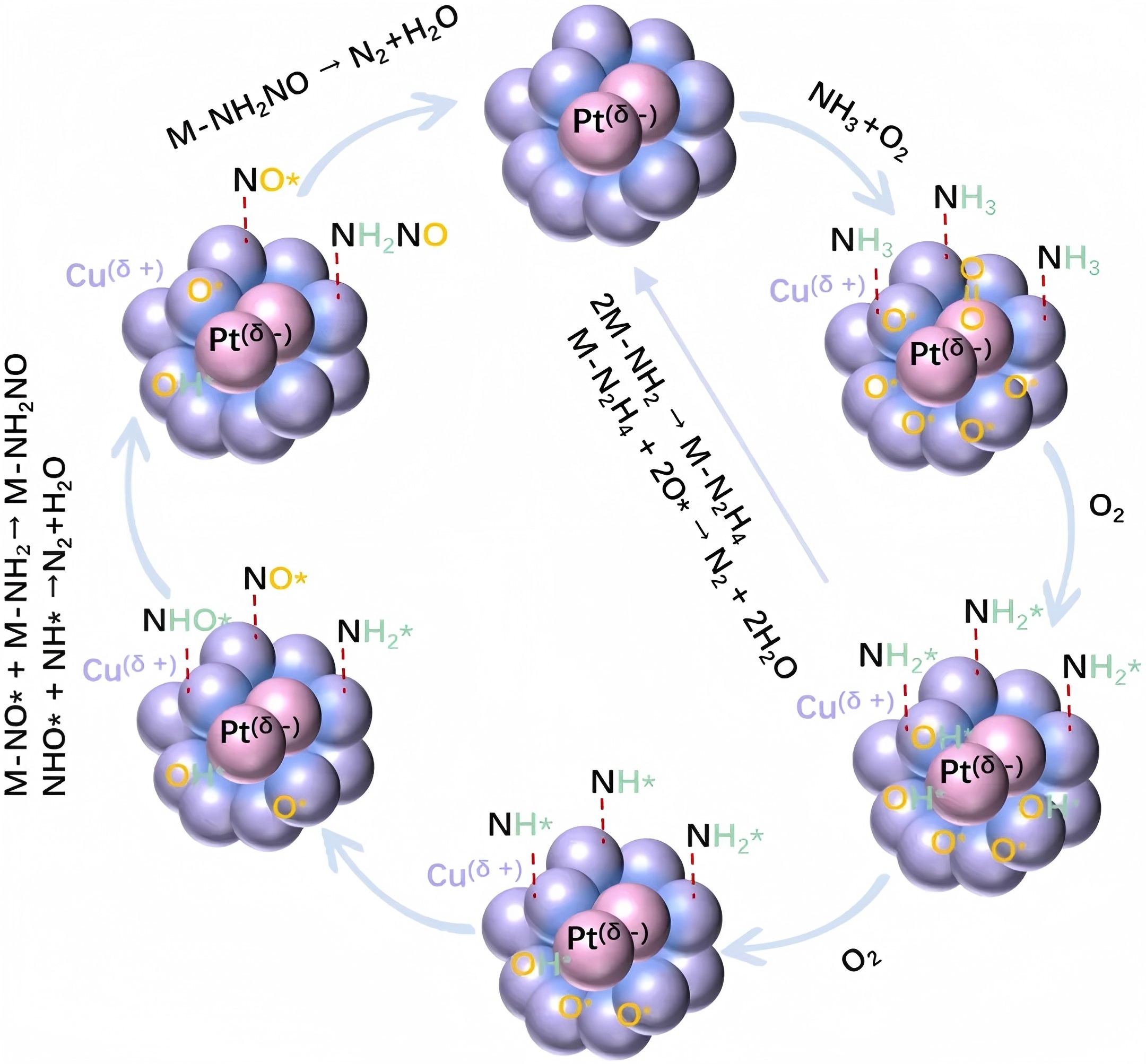
该研究成功构建了分子筛限域的PtCu双金属亚纳米团簇体系,通过原子级空间限域与界面电子调控的耦合策略,有效解决了NH3-SCO反应中活性与选择性的权衡难题。PtCu@S-1催化剂的优异性能源于三方面机制:其一,分子筛孔道限制金属迁移,抑制高温烧结,确保活性位点长期稳定;其二,紧密接触的PtCu界面诱发电子转移,形成极化活性位,实现氧气活化与氨吸附的功能分立与协同;其三,限域环境稳定了反应中间体,引导反应沿高选择性路径进行。该成果不仅提供了兼具高活性、高选择性和高稳定性的氨氧化催化剂实用设计方案,更揭示了限域体系中电子效应与几何效应的协同机制,为环境催化与能源转化领域双金属催化剂理性设计提供了可迁移的方法论框架。研究证实,原子尺度限域结构能够同步优化电子结构与空间构型,代表了多相催化设计的重要发展方向。